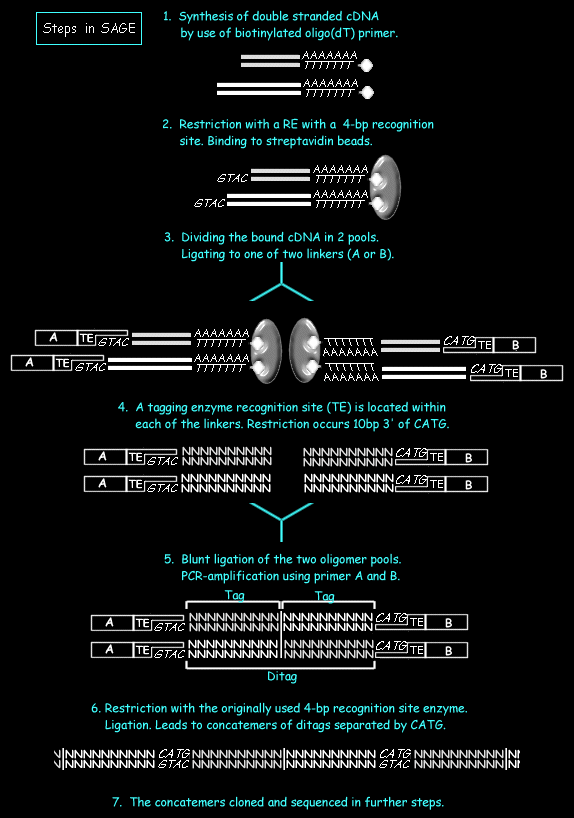
- Messenger RNA is harvested from the experimental tissue. A double stranded cDNA is synthesized from mRNA of the tissue or cells by using a biotinylated oligo(dT) primer.
- The obtained cDNA is digested with a ferquently cutting restriction enzyme, the anchoring enzyme(AE), with a 4-bp recognition site (as NlaIII, Sau3A). The most popular NlaIII will be considered in the further. Using the biotin label the 3'-most digestion products are captured restricting the study to the restriction site closest to the Poly-A site.
- The bound cDNA has to be separated in two pools. In both pools a ligation reaction is performed attaching a linker of either of two types to the 3' overhang of the digested cDNA. From 5' to 3' the linker contains a primer sequence of either type A or B, a recognition site for a type IIS restriction endonucleases (the tagging enzyme, TE), and a recognition site for the AE. The nature of the TE will determine the length of the tag sequences obtained. Short (10 base) nucleotide sequences are often sufficiently specific to discriminate different transcripts from each other. The choice of the length of the transcript is based on the estimation that a 9-base pair sequence can distinguish 262,144, with human genes being estimated to be below 100,000.
- The reaction is digested with the TE, which cleaves DNA at a position 14-18 base pair (dependent on the enzyme chosen) from its recognition site. This releases the linker along with short tags from a defined position within each of the mRNA transcripts, namely 3' of the AE restriction site closest to the Poly-A site.
- The two pools of tag-linker oligonucleotides are unified again, ligated and subjected to a PCR reaction. The amplified products contain two tags (ditags) bordered by linker DNA.
- &8. The linker DNA is removed from the ditags by a AE-digest. The obtained fragments are ligated together to form a concatemer of tags which are cloned into a standard plasmid vector and sequenced. Concatenated arrays are combined to form SAGE "libraries". The number of times a particular tag is detected in a library gives a digital measure of the abundance of its associated mRNA and, hence, provides a quantitative measure of gene expression. After collecting SAGE libraries the corresponding mRNA identity of each tag sequence must be determined. Several on-line SAGE tag database can be used that allow the researcher to match a tag to transcript:
- http://www.ncbi.nlm.nih.gov/SAGE
The NIH SAGE tag database provides human mRNA sequence data specific to the SAGE techniques as well as general descriptive information about the matching Unigen-cluster, mRNA, proteins. SAGEmap provides tag-to-gene as well as gene-to-tag mappings. Both mappings are updated weekly, immediately following the updating of UniGene. - http://sciencepark.mdanderson.org/ggeg/search.htm
The GGEG database provides human mRNA sequence data specific to the SAGE techniques as well as general descriptive information about the mRNAs. Sequence data is collected from the NCBI Entrez and Unigene databases and processed to extract the SAGE data. - High trough put by reducing transcription sequence information to a short tag.
- Highly sensitive, by scoring singular events.
- Quantitative measure obtained.
- Immortalized data allows for multiple comparisons.
- Avoids amplification bias.
- Includes unknown genes (eg. TEM2, TEM5, TEM9, TEM16, TEM22 etc.).
- Transcripts that are unlikely to be represented in EST databases can be analysed, as the method is unbiased.
- Loss of fidelity due to the conversion of a transcript to a short sequence tag is observed. One transcript can be described by more than just one tag (due to Poly-A independet tag amplification or alternative termination sites, eg. tem10_20, tem23_38, tem8_pem68, tem42_pem24), additionally one tag might occur in different transcripts (tem21, tem31).
- Data acquisition problems: Sequencing error have an increasing impact due to the shortness of the sequence information and the fact that singular events are evaluated.
- Limited information on alternative transcripts.
 |
 |
 |
| Home | Initium | Angiogenesis | FAQ |
| The SAGE - technique |
|---|
A short description of the SAGE - technique
Serial Analysis of Gene Expression (SAGE) is a high through put technique for measuring gene expression profiles (Velculescu et al, 1995). The Technology has been developed by Bert Vogelstein, Ken Kinzler, and Victor Velculescu at the Johns Hopkins University Oncology Center. Briefly, short cDNA tags (10-14bp) corresponding to unique transcripts are isolated and concatenated into single molecules, allowing for the simultaneous sequencing of thousands of tags. The relative tag abundance estimated in the sequencing step reflects the transcription patterns of the corresponding genes, allowing differential analysis of expression levels.
The steps of a SAGE- experiment are as follows (see the following figure):
| Advantages of SAGE: | Disadvantages of SAGE: |
|---|---|
Reference
Velculescu, V.E., Zhang, L., Vogelstein, B., and Kinzler, K.W. (1995) Science, 270, 484-487.