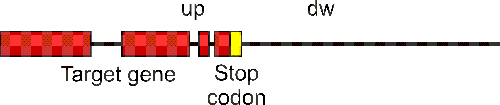
The target gene's flanking regions upstream of the start codon and downstream of the stop codon are designated "up" and "dw", respectively.
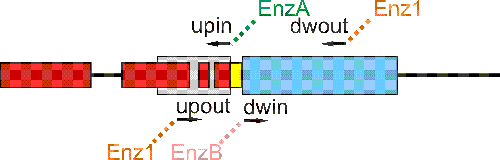
PCR primers are picked in silico so that 200 to 800 bp of up and dw regions can be amplified. The primers distal to the target gene ("upout" and "dwout") contain 5'-tails encoding a restriction site for the same enzyme "Enz1". The proximal primers ("upin" and "dwin") contain 5'-tails enconding restrictions site for two different enzymes, "EnzA" and "EnzB".
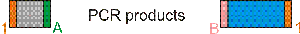
The PCR products are mixed and purified using a column. T4 ligase is added and amplified homology regions are ligated. Of all possible ligation products, the desired ones are heterodimers of the two homology regions in inverted orientation. After incubation, the DNA is purified using a column. The ligation products are incubated with enzymes EnzA and EnzB to create sticky ends for ligation into the vector and to resolve potential multimers that were created during the ligation into dimers.
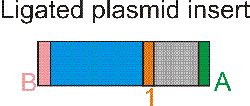
The reaction is run on an agarose gel. Because PCR primers are picked so that up and dw regions are of unequal size, usually five bands should be seen. These correspond to (from lower to higher weight) the smaller monomer, the larger monomer, the homodimer of the smaller monomer, the desired heterodimer and the homodimer of the larger monomer. The heterodimer band is excised from the gel and DNA is extracted using a column. The purified heterodimer is ligated to a vector cut with enzymes EnzA and EnzB and bacteria are transformed.
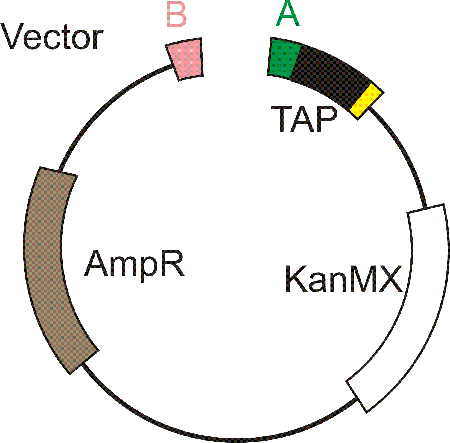
Transformant colonies are checked for integration of the correct target by colony PCR using two primers "upch-uni" and "dwch-uni" (see below). After preparation of the plasmid from bacteria, it is digested with enzyme Enz1, the same enyzme that was used to ligate the two monomers together. The linearised plasmid is transformed into yeast (S. pombe) cells which are plated out on selective medium.
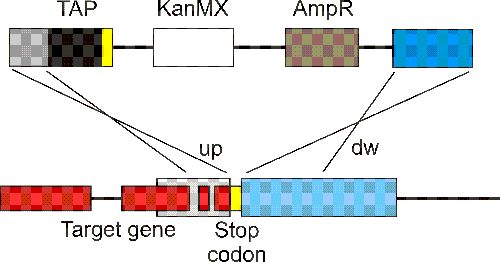
After yeast colonies are formed, clones are checked for correct integration of the tagging construct by PCR using two primers specific for the target gene ("upch" and "dwch") and two primers specific for the plasmid ("upch-uni" and "dwch-uni"). Two PCR reactions test for correct integration in up and dw regions.