| |
||
ProflAP - A superfamily of profilin-like adaptor proteins
This page is an extended supplementary material accompanying the article:
Kurzbauer R, Teis D, de
Araujo ME, Maurer-Stroh S, Eisenhaber F, Bourenkov GP, Bartunik HD, Hekman M, Rapp UR, Huber LA, Clausen T.
Crystal structure of the p14/MP1 scaffolding complex: how a twin couple
attaches mitogen-activated protein kinase signaling to late endosomes.
Proc Natl Acad Sci U S A. 2004 Jul 27;101(30):10984-9.
Article - http://www.pnas.org/cgi/content/full/101/30/10984
Compact supplement - http://www.pnas.org/cgi/content/full/0403435101/DC1
It is divided into the following subsections:
Extension of a sequence superfamily and multiple sequence alignment
Functional implications of sequence superfamily relationships
Functional implications of structure superfamily relationships
Extension of a sequence superfamily
p14 was already known to be part of an ancient superfamily (ref.)
that extends from a eukaryotic family of Roadblock dynein light chains (KOG4115) to several archaeal and bacterial
proteins that were summarized as MglB-family (COG2018). Progress of sequencing projects has increased the searchable
protein sequence space, which allows the detection of further homology relationships between proteins and protein
families. Extensive iterative sequence searches with the PSI-BLAST (ref.)
program starting with all members of the superfamily, as previously defined, against the non-redundant database at
the National Center for Biotechnology (NCBI), establish significant connections of the MglB-family not only with the
large family of Profilins (KOG1755) but also with the p14 interaction partner MP1 (for details see Methods section).
It has been noted that conserved predicted secondary structural elements among the family members would point to a
conserved fold (ref.)
which also applies to the extended superfamily (with the exception of the Roadblock dynein light chains that
apparently lack the C-terminal helix). The linkage of the protein families by sequence similarity is further
strengthened by similarity on the structural level that is indicated by the strong correspondence of the folds of
Profilins with those of p14 and MP1 that are described in this work. We therefore extend the superfamily to include
the following families: KOG4115 (Roadblock, etc.), COG2018 (MglB, etc.), KOG4107 (p14), KOG1755 (Profilins) and MP1s
(no KOG at time of publication). It is interesting to note that plant Profilins represent the first hits on the
sequence level with bacterial members of the superfamily and also appear to structurally more closely resemble the
fold of p14 and MP1. Below follows a multiple alignment of the ProflAP superfamily sequences and the three available
structures (for details see Methods section):
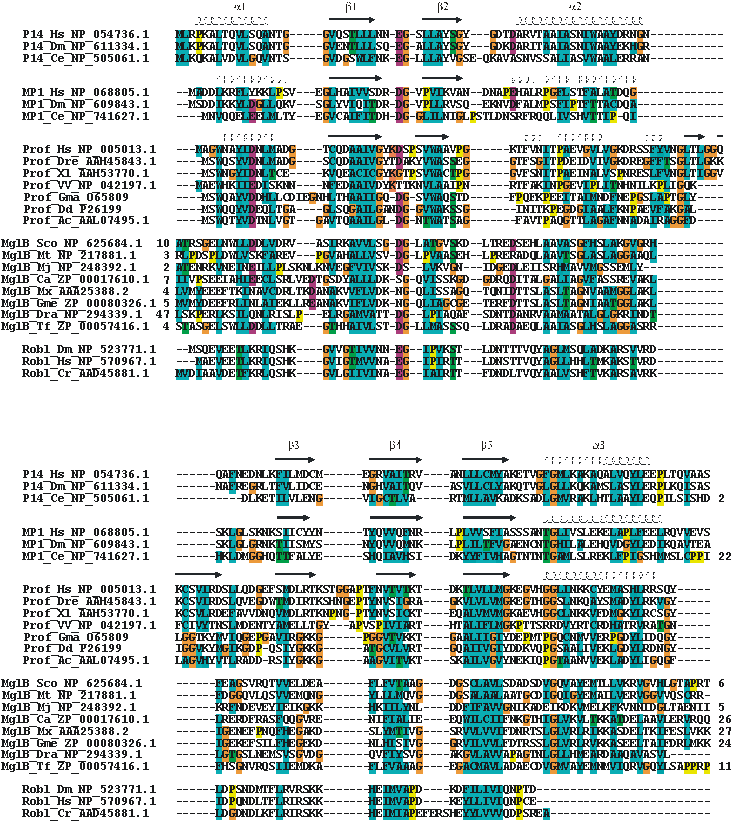
Multiple alignment of the ProflAP superfamily.
Functional implications of sequence
superfamily relationships
Several proteins of the MglB-family appear in the genetic context, for example in the same operon, with a GTPase from
the Ras/Rab/Rho superfamily. Dynein light chains form complexes with the dynein heavy chains that consist of ATPases.
Hence, a common role for the superfamily in NTPase regulation has been suggested (ref.).
The context of NTPase interaction is also fulfilled indirectly by Profilins which are regulated by Ras/Rab/Rho
GTPases through interaction with other adaptor proteins. For example N-WASP binds both Profilin and the GTPase Cdc42
(ref.).
Another common feature of several superfamily members appears to be a connection to subcellular scaffold structures.
Roadblock dynein light chains form part of the complex that connects cargo to dynein motors to be transported along
microtubules (ref.).
Profilin is involved in regulating actin polymerization and rearrangment of the actin cytoskeleton plays a role in
eukaryotic cell motility (ref.).
Mutations in MglB and the neighboring GTPase MglA affect gliding motility in Myxococcus xanthus. Although the
mechanisms in bacteria behind the general term ‘gliding’ are diverse, they are often involving
subcellular scaffold structures (ref.).
With the extension of the superfamily it becomes clear that the functions of the different proteins might cover a
much broader spectrum. For example, a gene for a helix-turn-helix (HTH) protein (corresponding to domain of unknown
function DUF742) appears conserved inbetween the GTPase and MglB-family genes in the genomes of at least 6 different
actinobacteria (Mycobacterium tuberculosis, Mycobacterium bovis, Streptomyces coelicolor, Streptomyces griseus,
Streptomyces avermitilis, Thermobifida fusca). Furthermore, there exists a structure of a family of bacterial
transcriptional regulator proteins (IclR-family = COG1414) consisting of an N-terminal HTH- and a C-terminal
Profilin-fold domain (ref.).
No significant sequence similarity is currently found for the Profilin-fold domain with the superfamily described
here, but a significant sequence link can be made between DUF742 and IclR-family members (for details see Materials
and Methods section). This relationship would be in favor of a gene fusion of DUF742 (HTH-fold) and the MglB-family
(Profilin-fold) neighboring genes resulting in a domain arrangement as in the IclR-family (HTH-fold+Profilin-fold).
This example adds transcriptional regulation as yet another facet to the possible functions that could be related to
members of the superfamily.
Functional implications of structure
superfamily relationships
The structure of Profilin, among proteins with resolved structure most closely related to p14 and MP1, shows some
characteristic differences, mainly in the region building up the binding interface of the p14/MP1 complex. Loop b3
appears extended and helix b translocated along the helix axis while additional residues between helix b and beta
strand 3 form a complex 30 amino acid long alpha-beta mixed scaffold that also builds up parts of the large area
actin binding site (ref.).
Aromatic residues intercalating between helix a and c form the binding groove for proline-rich peptides in Profilins
(ref.).
Furthermore, clusters of positive charges on the surface are likely sites of interactions with PIP2 (ref.).
The latter feature, of PIP2 binding over positively charged surface regions, could be conserved to the p14/MP1
complex and influence its subcellular localization by targeting to membranes.
Besides the members of the sequence superfamily, there are also several more proteins that share similarities to the
fold of Profilin, p14 and MP1 as unveiled by searches against the PDB database using the DALI structure comparison
tool (ref.).
The fact that we cannot find significant sequence similarities between the structurally related Profilin-fold domain
of IclR transcriptional regulator with the sequence superfamily, although the genetic context would suggest an
evolutionary relationship, nurtures the idea that also other structural neighbors could share more than just the
basic scaffold. Of course, molecular and cellular function conservation of proteins that appear to be related merely
through structural similarities vanishes during the aeons of evolution. However, it is interesting that in the IclR
structure the Zn2+ signal for transcriptional regulation is presumed to bind to a site (ref.)
whose location corresponds to the center of the dimerization interface of the p14/MP1 complex. As p14 and MP1 act as
adaptors to localize MAPK signaling to late endosomes, several proteins that share the Profilin-fold and are also
related to endosomal functions or localizations have caught our attention. For example, vesicle trafficking protein
Sec22b, the N-terminal domain of nonsyntaxin SNARE protein Ykt6, transport protein particle component SEDL and
adaptor-related protein complex 2 (AP2) subunits alpha-2 and N-mu-2, all belong to the Profilin-like fold family. It
is striking, however, that all of these structures are circular permutations of the original Profilin fold breaking
loop b1 and connecting helices a and c. Therefore, helix a appears at the C- instead of the N-terminus in the
sequences.
Similarily to p14 and MP1, Sec22b has conserved surface residues in the b3-pocket, loop b2* and conserved hydrophobic
residues in the region corresponding to the p14/MP1 dimerization interface (ref.).
As biochemical data does not support homodimerization of the protein, a heterodimerization in a similar style as in
the p14/MP1 complex or other binding partner interactions of this region are plausible. In this context it is
interesting that the mammalian Roadblock dynein light chains Robl1 and Robl2 that are predicted to share the fold of
the superfamily have been found to be capable of forming both homo- and heterodimers (ref.).
Ykt6, also features a conserved stretch of hydrophobic residues in the region corresponding to the p14/MP1
dimerization interface, which is presumed to be required for regulatory interaction with its C-terminal SNARE core
domain (ref.).
A similar conserved mainly hydrophobic patch can be found in the SEDL structure and it is interesting to note that a
mutation responsible for the X-linked skeletal disorder spondyloepiphyseal dysplasia tarda maps to this surface
region (ref.).
Additionally, there appears another potential solvent-exposed hydrophobic protein interaction site in the loop that
is formed between helices a and c (p14 nomenclature) after the circular permutation, which in the structural
alignment would correspond exactly to the conserved site where the N-terminus of p14 tries to bridge the gap to the
flexible but conserved C-terminus. On the contrary, the AP2 subunits alpha-2 and N-mu-2 do not appear to share
conserved interaction locations with p14 and MP1 as they seem to undergo more large-area interactions with other
subunits and perform a scaffolding role in the AP2 complex (ref.).
The pattern of binding sites for diverse interaction partners located around the center of the interaction interface
between p14 and MP1 continues with other more distantly related proteins sharing the Profilin fold but do not have
the circular permutation as the previous examples. For instance, the GAF domains of 3’,5’-cyclic
nucleotide phosphodiesterase 2a bind cGMP in this location that is furthermore embraced by an extended loop between
adjacent beta sheets (ref.).
More hidden by an additional mainly helical scaffold are the active or binding sites of d-Ala carboxypeptidase,
beta-lactamase and penicillin-binding proteins (ref.).
Another structural neighbor, that shares the circular permutation with the SNARE related proteins mentioned above, is
the signal recognition particle receptor alpha subunit homolog. The fold similarity is of special interest since the
SRX domain of the alpha subunit is an effector for the beta subunit which is a GTPase (ref.).
Moreover, the interaction site of the GTPase with the Profilin-fold protein corresponds to the b2 loop that in most
members of the superfamily has a conserved negative charge followed by a glycine and has been proposed to be involved
in GTPase regulation (ref.).
On the other hand, the fact that this loop is conserved also in several more of the only structurally related
proteins with GTPase independent functions could point out that its role might lie more in the stabilization of the
structure in this region.
Extension of the ProflAP superfamily:
A fan-like search strategy
(ref.)
using PSI-BLAST (ref.)
was applied to find new superfamily members in the non-redundant database (NR) at the National Center for
Biotechnology (NCBI). Accession numbers of representative pairs linking families as well as their expectation values
(E-value) at first encounter in the PSI-BLAST procedure are listed below. COG2018 (MglB-family) finds KOG1755
(Profilins): ZP_00057416.1 (residues 11 to 126; Thermobifida fusca) hits O65809 (residues 1 to 126; Glycine max) in
round 1 with an E-value of 0.003. COG2018 (MglB-family) finds MP1s: NP_294339.1 (residues 56 to 166; Deinococcus
radiodurans) hits AAP06349.1 (residues 7 to 124; Schistosoma japonicum) in round 5 with an E-value of 0.002.
Helix-turn-helix (HTH) motif of DUF742 finds HTH motif of COG1414 (IclR): NP_627109.1 (residues 49 to 107;
Streptomyces coelicolor) hits ZP_00019631.1 (residues 44 to 102; Chloroflexus aurantiacus) in round 3 with an E-value
of 0.002.
Multiple alignment of the ProflAP superfamily:
Selected sequences were aligned to a profile derived from a structural alignment of p14, MP1 and profilin (secondary structures on top of the corresponding sequences) using ClustalX (ref.) and setting secondary structure specific gap penalties. Family, organism and accession numbers are given. Numbers preceding or consecutive to sequences denote residue extensions. Standard ClustalX coloring (shortly: hydrophobic – cyan, polar – green, negative – red background). Abbreviations: Prof – profilin family, MglB – archeal or bacterial MglB-family member, Robl – Roadblock/LC7 family; Hs – Homo sapiens, Dm – Drosophila melanogaster, Ce – Caenorhabditis elegans, Dre – Danio rerio, Xl –Xenopus laevis, VV – Variola virus, Gma – Glycine max, Dd - Dictyostelium discoideum, Ac - Acanthamoeba castellanii, Sco - Streptomyces coelicolor A3(2), Mt - Mycobacterium tuberculosis H37Rv, Mj - Methanococcus jannaschii, Ca - Chloroflexus aurantiacus, Mx - Myxococcus xanthus, Gme - Geobacter metallireducens, Dra - Deinococcus radiodurans, Tf - Thermobifida fusca, Cr - Chlamydomonas reinhardtii.
Multiple alignment of the ProflAP superfamily (Clustal format)
Calibrated Hidden Markov Model of the ProflAP superfamily (HMMer format)